La malattia prende il nome dalla regione geografica in cui è più diffusa, il Mediterraneo, ma è presente in tutto il mondo, influenzando diverse popolazioni.
Eziologia e Genetica
La talassemia è una malattia genetica autosomica recessiva, il che significa che per sviluppare la malattia un individuo deve ereditare una copia mutata del gene della talassemia da entrambi i genitori (Cao & Galanello, 2010, Orphanet Journal of Rare Diseases). La malattia è causata da mutazioni nel DNA che interferiscono con la produzione normale di emoglobina. Le mutazioni possono influenzare uno di due geni, il gene HBA, responsabile della produzione delle catene alfa dell’emoglobina, o il gene HBB, responsabile delle catene beta (Steinberg & Forget, 2001, NEJM).
Tipologie e Sintomi
Ci sono due principali tipi di talassemia: alfa e beta, a seconda della catena dell’emoglobina interessata dalla mutazione genetica. La gravità della malattia varia a seconda del numero di geni mutati (Weatherall, 2001, Nature).
La talassemia alfa può variare da asintomatica a grave; nei casi più gravi, può provocare morte fetale o neonatale. I sintomi nei casi meno gravi includono ittero, pallore, crescita lenta, spossatezza e deformità ossee facciali (Galanello & Cao, 2011, GeneReviews).
La talassemia beta può essere suddivisa in minore (o trait), intermedia e maggiore. La talassemia beta minore è spesso asintomatica, mentre la forma intermedia provoca sintomi di anemia di gravità variabile. La talassemia beta maggiore, o anemia di Cooley, è la forma più grave e può causare sintomi gravi, incluso sviluppo fisico lento, pallore, ittero, ingrossamento della milza e deformità ossee (Origa, 2017, GeneReviews).
Diagnosi e Trattamento
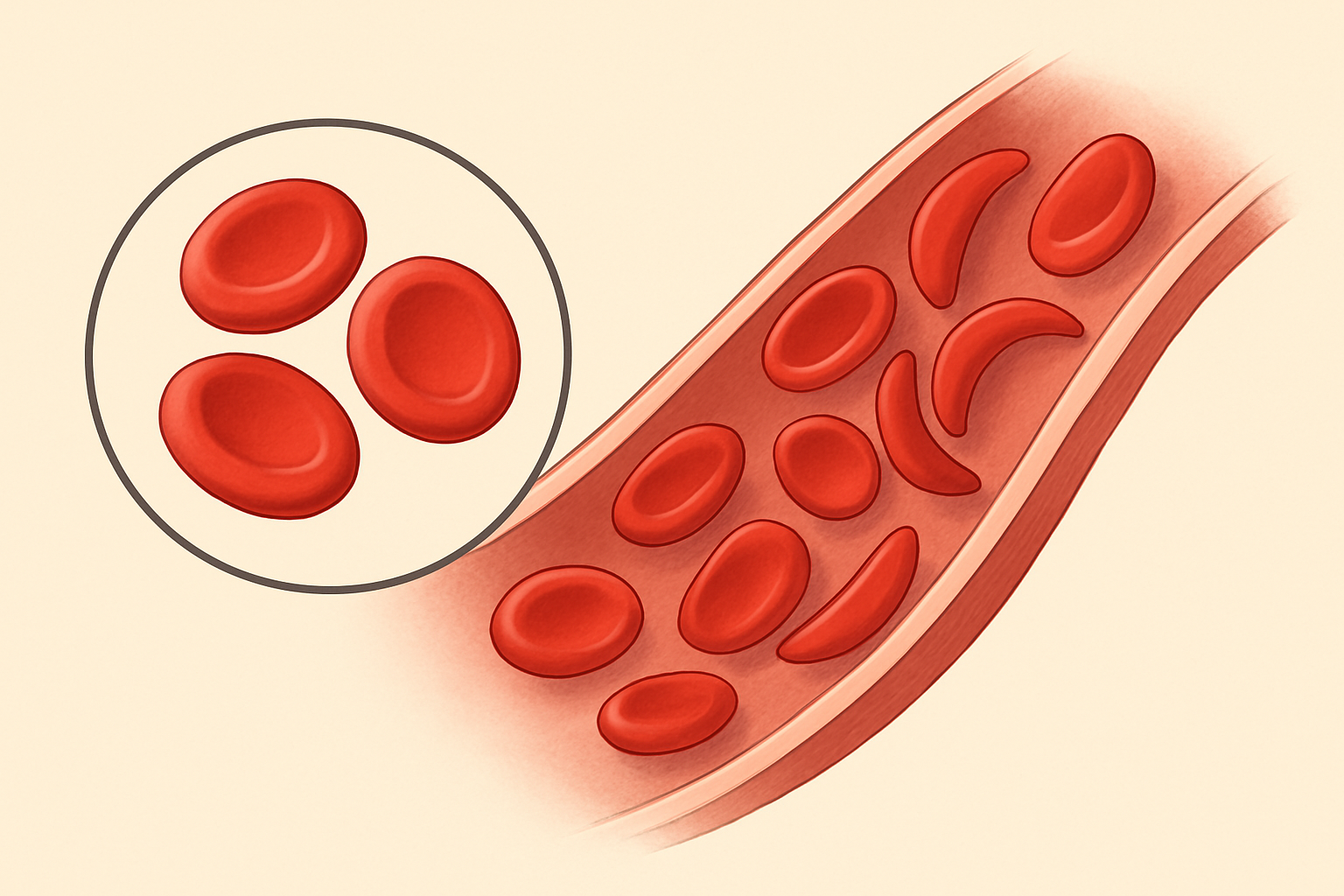
La diagnosi di talassemia si basa su test del sangue che mostrano anemia e globuli rossi piccoli e malformati, e può essere confermata da test genetici (Weatherall, 2010, Lancet).
Il trattamento della talassemia varia a seconda della gravità della malattia. Le opzioni di trattamento includono trasfusioni di sangue regolari, farmaci per ridurre l’accumulo di ferro nel corpo (chelanti del ferro), acido folico supplementare e, nei casi più gravi, trapianto di cellule staminali ematopoietiche (Lucarelli & Gaziev, 2012, Haematologica).
Ricerca e Prospettive Future
La ricerca sulla talassemia sta esplorando nuovi modi per trattare e prevenire questa malattia. Tra questi, la terapia genica, che mira a correggere o sostituire il gene difettoso, sta mostrando risultati promettenti (Thompson et al., 2018, Nature Medicine). Inoltre, studi su farmaci che aumentano la produzione di emoglobina fetale, una forma naturale di emoglobina che può compensare la carenza di emoglobina normale, stanno procedendo (Sankaran et al., 2008, Science).
In conclusione, l’anemia mediterranea è una malattia genetica complessa con una vasta gamma di presentazioni cliniche. Mentre i progressi nel trattamento hanno migliorato notevolmente la prognosi per molte persone con talassemia, la ricerca continua per trovare cure più efficaci e forse un giorno una cura definitiva.